




How will peptide deconvolution affect my identifications?
To quantify using Hi-N, Progenesis needs to perform peptide deconvolution. For new experiments, this is performed automatically after importing identifications.
If you open an experiment saved in an older version of Progenesis, and choose to quantify using Hi-N, Progenesis will ask your permission to perform peptide deconvolution retrospectively:
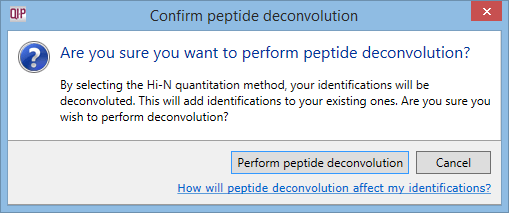
The dialog shown when selecting Hi-N in experiments saved with an older version of Progenesis.
This FAQ explains what deconvolution is, why it is needed for Hi-N quantitation, and how deconvolution will affect your existing identifications.
What is peptide deconvolution?
Ionization produces various charge states of the same peptide, but it may be that your chosen search engine can only identify a subset of these. Peptide deconvolution shares the search engine identifications across different charge states of the same peptide.
For each search engine identification, Progenesis calculates where other charge states of the same peptide would be expected. For each of these locations, if a peptide ion exists there with a similar elution profile, it is assigned the same identification. For more information, see How does peptide ion deconvolution work?
Why is peptide deconvolution necessary for Hi-N?
When performing absolute quantitation via Hi-N, it is important to use an accurate measure of total peptide abundance. Peptide deconvolution ensures this by summing the abundance of all charge states of a given peptide.
How will peptide deconvolution affect my identifications?
Progenesis QI will add an identification to any peptide ion it detects as a different charge state of a search engine identified peptide. No search engine identifications will be deleted or modified.
Any identifications resulting from a previous deconvolution will be removed before re-performing deconvolution.
Deconvolution example
In the example below, the charge 2 peptide ion at m/z 539.3004 was identified as LLFTQVDNK by the search engine. The charge 1 ion at m/z 1077.5933 was not identified by the search engine.
Progenesis calculated that a charge 2 peptide ion (shown in red) with m/z 539.3004 must have a neutral peptide mass of 1076.586, therefore a charge 1 ion would appear at m/z 1077.5933.
There is a charge 1 ion at this m/z (shown in blue), and its elution profile is similar, so Progenesis assumes it is a charge 1 state of LLFTQVDNK. It is therefore assigned the same peptide sequence, but with a --- for its Peptide Score.
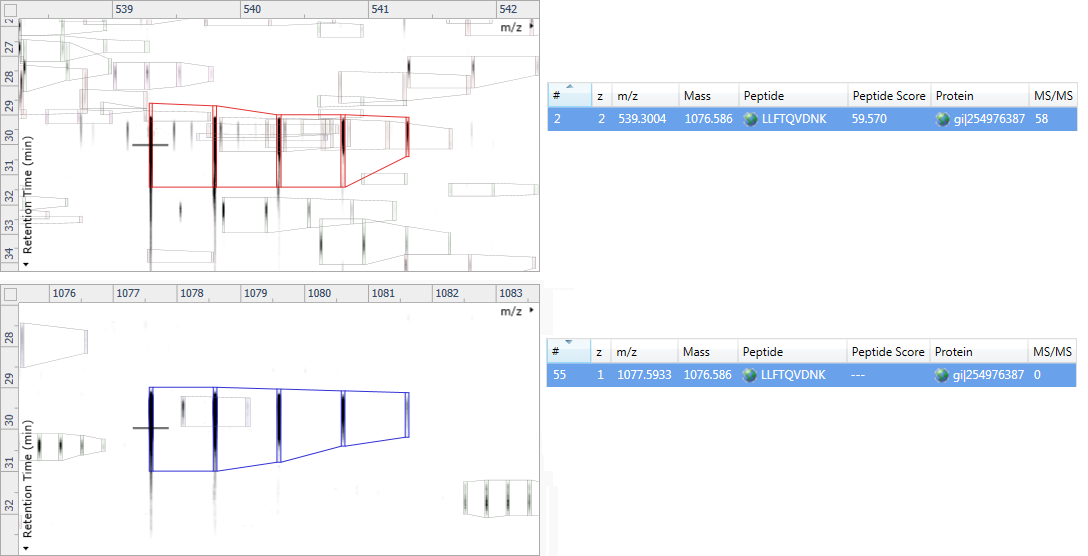
An example of peptide deconvolution. The charge 2 ion was identified by the search engine as LLFTQVDNK, and the charge 1 ion was assigned this ID via deconvolution.