





P-values, False Discovery Rate (FDR) and q-values
What are p-values?
The object of differential analysis is to find those compounds that show abundance difference between experiment groups, thereby signifying that they may be involved in some biological process of interest to the researcher. Due to chance, there will always be some difference in abundance between groups. However, it is the size of this difference in comparison to the variance (i.e. the range over which abundance values fall) that will tell us if this abundance difference is significant or not. Thus, if the difference is large but the variance is also large, then the difference may not be significant. On the other hand, a small difference coupled with a very small variance could be significant. We use Anova tests to formalise this calculation. The tests return a p-value that takes into account the mean difference and the variance and also the sample size. The p-value is a measure of how likely you are to get this compound data if no real difference existed. Therefore, a small p-value indicates that there is a small chance of getting this data if no real difference existed and therefore you decide that the difference in group abundance data is significant. By small we usually mean a probability of 0.05.
What are q-values, and why are they important?
False positives
A positive is a significant result, i.e. the p-value is less than your cut off value, normally 0.05. A false positive is when you get a significant difference where, in reality, none exists. As I mentioned above, the p-value is the chance that this data could occur given no difference actually exists. So, choosing a cut off of 0.05 means there is a 5% chance that we make the wrong decision.
The multiple testing problem
When we set a p-value threshold of, for example, 0.05, we are saying that there is a 5% chance that the result is a false positive. In other words, although we have found a statistically significant result, there is, in reality, no difference in the group means. While 5% is acceptable for one test, if we do lots of tests on the data, then this 5% can result in a large number of false positives. For example, if there are 2000 compounds in an experiment and we apply an Anova or t-test to each, then we would expect to get 100 (i.e. 5%) false positives by chance alone. This is known as the multiple testing problem.
Multiple testing and the False Discovery Rate
While there are a number of approaches to overcoming the problems due to multiple testing, they all attempt to assign an adjusted p-value to each test or reduce the p-value threshold from 5% to a more reasonable value. Many traditional techniques such as the Bonferroni correction are too conservative in the sense that while they reduce the number of false positives, they also reduce the number of true discoveries. The False Discovery Rate approach is a more recent development. This approach also determines adjusted p-values for each test. However, it controls the number of false discoveries in those tests that result in a discovery (i.e. a significant result). Because of this, it is less conservative that the Bonferroni approach and has greater ability (i.e. power) to find truly significant results.
Another way to look at the difference is that a p-value of 0.05 implies that 5% of all tests will result in false positives. An FDR adjusted p-value (or q-value) of 0.05 implies that 5% of significant tests will result in false positives. The latter will result in fewer false positives.
q-values
Q-values (Storey and Tibshirani 2003) are the name given to the adjusted p-values found using an optimised FDR approach. The FDR approach is optimised by using characteristics of the p-value distribution to produce a list of q-values. In what follows, I will tie up some ideas and hopefully this will help clarify what we have been saying about p and q values.
It is usual to test many hundreds or thousands of compound variables in a metabolomics experiment. Each of these tests will produce a p-value. The p-values take on a value between 0 and 1 and we can create a histogram to get an idea of how the p-values are distributed between 0 and 1. Some typical p-value distributions are shown below. On the x-axis, we have histogram bars representing p-values. Each bar has a width of 0.05 and so in the first bar (red or green) we have those p-values that are between 0 and 0.05. Similarly, the last bar represents those p-values between 0.95 and 1.0, and so on. The height of each bar gives an indication of how many values are in the bar. This is called a density distribution because the area of all the bars always adds up to 1. Although the two distributions appear quite different, you will notice that they flatten off towards the right of the histogram. The red (or green) bar represents the significant values, if you set a p-value threshold of 0.05.
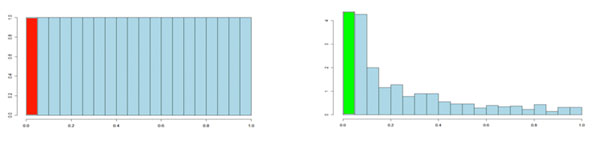
If there are no significant changes in the experiment, you will expect to see a distribution more like that on the left above while an experiment with significant changes will look more like that on the right. So, even if there are no significant changes in the experiment, we still expect, by chance, to get p-values < 0.05. These are false positives, and shown in red. Even in an experiment with significant changes (in green), we are still unsure if a p-value < 0.05 represents a true discovery or a false positive. Now, the q-value approach tries to find the height where the p-value distribution flattens out and incorporates this height value into the calculation of FDR adjusted p-values. We can see this in the histogram below. This approach helps to establish just how many of the significant values are actually false positives (the red portion of the green bar).
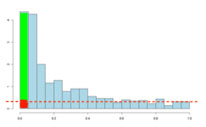
Now, the q-values are simply a set of values that will lie between 0 and 1. Also, if you order the p-values used to calculate the q-values, then the q-values will also be ordered. This can be seen in the following screen shot from Progenesis QI; notice that q-values can be repeated:
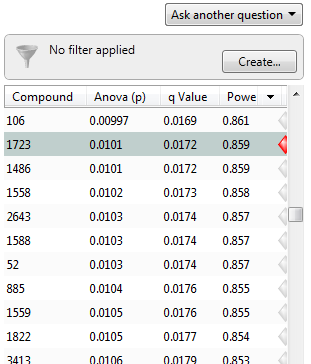
To interpret the q-values, you need to look at the ordered list of q-values. There are 3516 compounds in this experiment. If we take unknown compound 1723 as an example, we see that it has a p-value of 0.0101 and a q-value of 0.0172. Recall that a p-value of 0.0101 implies a 1.01% chance of false positives, and so with 3516 compounds, we expect about 36 false positives, i.e. 3516 × 0.0101 = 35.51. In this experiment, there are 800 compounds with a value of 0.0101 or less, and so 36 of these will be false positives.
On the other hand, the q-value is 0.0172, which means we should expect 1.72% of all the compounds with q-value less than this to be false positives. This is a much better situation. We know that 800 compounds have a q-value of 0.0172 or less and so we should expect 800 × 0.0172 = 13.76 false positives rather than the predicted 36. Just to reiterate, false positives according to p-values take all 3516 values into account when determining how many false positives we should expect to see while q-values take into account only those tests with q-values less than the threshold we choose. Of course, it is not always the case that q-values will result in less false positives, but what we can say is that they give a far more accurate indication of the level of false positives for a given cut-off value.
When doing lots of tests, as in a metabolomics experiment, it is more intuitive to interpret p and q values by looking at the entire list of values in this way rather that looking at each one independently. In this way, a threshold of 0.05 has meaning across the entire experiment. When deciding on a cut-off or threshold value, you should do this from the point of view of how many false positives will this result in, rather than just randomly picking a p- or q-value of 0.05 and saying that every compound with p-value less this is significant.
References
- Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. PNAS 100 (16):9440-9445