






What is the correct format for compounds in SDF or MOL files?
Molfiles are text files which contain structure information for a single molecular compound. SDFs (structure data files) consist of a series of molfiles joined together, together with some additional information about the compounds. They are frequently used for sharing libraries of compound structure data.
702
-OEChem-02271511112D
9 8 0 0 0 0 0 0 0999 V2000
0.5369 0.9749 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
1.4030 0.4749 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.2690 0.9749 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.8015 0.0000 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1.0044 0.0000 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1.9590 1.5118 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
2.8059 1.2849 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
2.5790 0.4380 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
0.0000 0.6649 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0 0 0 0
1 9 1 0 0 0 0
2 3 1 0 0 0 0
2 4 1 0 0 0 0
2 5 1 0 0 0 0
3 6 1 0 0 0 0
3 7 1 0 0 0 0
3 8 1 0 0 0 0
M END
> <ID>
00001
> <DESCRIPTION>
Solvent produced by yeast-based fermentation of sugars.
$$$$
A compound record contains several distinct sections. First, there is a three-line header block. These three lines may contain:
- The name of the molecule
- Details of the software used to generate the compound structure
- A comment
Alternatively, any (or all) of these lines may be left blank.
In the example above, the molecule's name is "702", was generated by "-OEChem-02271511112D", and its comment is blank.
The Counts line
Next comes the so-called "counts" line. This line is made up of twelve fixed-length fields - the first eleven are three characters long, and the last six characters long. The first two fields are the most critical, and give the number of atoms and bonds described in the compound.
9 8 0 0 0 0 0 0 0999 V2000
So this compound will have 9 atoms and 8 bonds described. Often, hydrogens - especially those attached to elements such as carbon or oxygen - are left implicit (and will be included based on the available valences) rather than being included in the file.
The Atoms block
After the counts line comes the atoms block. For each atom mentioned in the first field of counts, include a line like so:
0.5369 0.9749 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
The first three fields, 10 characters long each, describe the atom's position in the X, Y, and Z dimensions. After that there is a space, and three characters for an atomic symbol (O for oxygen, in this instance).
After the symbol, there are two characters for the mass difference from the monoisotope. This field only supports values between -3 and +4, however - the M ISO property can be used for values outside this range.
Next you have three characters for the charge. The values are a little confusing - see the conversion table. Alternatively, use the M CHG property instead, which is much less confusing and also supports a wider range of values.
| Charge | ||||||||
|---|---|---|---|---|---|---|---|---|
| You have a... | -3 | -2 | -1 | Neutral | +1 | +2 | +3 | Doublet radical |
| Written as... | 7 | 6 | 5 | 0 | 3 | 2 | 1 | 4 |
There are ten more fields with three characters each - but these are all rarely used, and can be left blank for the purposes of working with Progenesis SDF Studio or Progenesis MetaScope.
The Bonds block
After the atoms block, you next specify the bonds between them. Again, the length of this section will be determined by the number in the counts line.
1 2 1 0 0 0 0
The first two fields are the indexes of the atoms included in this bond (starting from 1). The third field defines the type of bond, and the fourth the stereoscopy of the bond.
| Bond type | Bond stereo | |||
|---|---|---|---|---|
| Value | Meaning | Value | Meaning | |
| 1 | Single | 0 | Not stereo | |
| 2 | Double | 1 | Up | |
| 3 | Triple | 6 | Down | |
| 4 | Aromatic | Other | See specification (zip) | |
| Other | See specification (zip) | |||
So the example means, "between the first and second atoms, add a single, non-stereo bond". There are a further three fields, with 3 characters each, but these are rarely used and can be left blank.
Properties
- Charge
M CHG 1 1 2
- After the M CHG, the first number defines the number of charges defined on this line (up to 8). If the compound has more than this, they can go on additional M CHG lines. Each charge entry consists of two four-character fields - the first is the index of the charged atom (starting from one), and the second is the charge. The above means "add a charge to the first atom of +2".
- Isotope
M ISO 1 1 2
- After the M ISO, the first number defines the number of isotopes defined on this line (up to 8). If the compound has more than this, they can go on additional M ISO lines. Each isotope entry consists of two four-character fields - the first is the index of the atom (starting from one), and the second is the atom's actual mass number. The above means "the first atom has an atomic mass of 2".
- Terminator
M END
- The M END property is compulsory, and must be included at the end of the properties list (whether or not there are any actual properties in it).
A number of other properties are defined in the specification (zip), and several programs implement custom properties of various types.
Data fields
A wide variety of custom metadata about a compound can be stored in data fields. Data fields start with a header, which begins with a >. On the same line, the name of the data field is written in angular brackets. The header line can contain other text too, though it is generally ignored.
> <ID> 00001
After the header, a data field contains one or more lines of up to 200 characters of free text, which is the value of the data field.
Progenesis SDF Studio will use any of the following fields as an identifier:
- PUBCHEM_COMPOUND_CID
- pubchem_compound_id
- PUBCHEM_SID
- PUBCHEM.external_id
- pubchem.external_id
- PUBCHEM_SUBSTANCE_ID
- LM_ID
- PdbId
- DATABASE_ID
- DRUGBANK_ID
- HMDB_ID
- hmdb_id
- HMDB.external_id
- DRUGBANK_ID
- DRUGBANK.external_id
- drugbank.external_id
- KEGG
- CAS
- CASNO
- CAS_RN
- ChEBI ID
- CAT_NO
- MDL_NO
- ID
and any of the following fields as a description:
- COMMON_NAME
- DRUGBANK_GENERIC_NAME
- LONGNAME
- Name
- GENERIC_NAME
- NAME
- HMDB_Name
- SYSTEMATIC_NAME
- IUPAC_NAME
- PUBCHEM_IUPAC_NAME
- JCHEM_IUPAC
- DRUGBANK_IUPAC_NAME
- TRADITIONAL_IUPAC_NAME
- iupac
- SMILES
- ChEBI Name
- DESCRIPTION
SDF separator ($$$$)
While it's not required in molfiles, the final line of each record in an SDF database contains only 4 dollar symbols ($$$$).
$$$$
Solving common problems in SDF databases
Missing lines from header
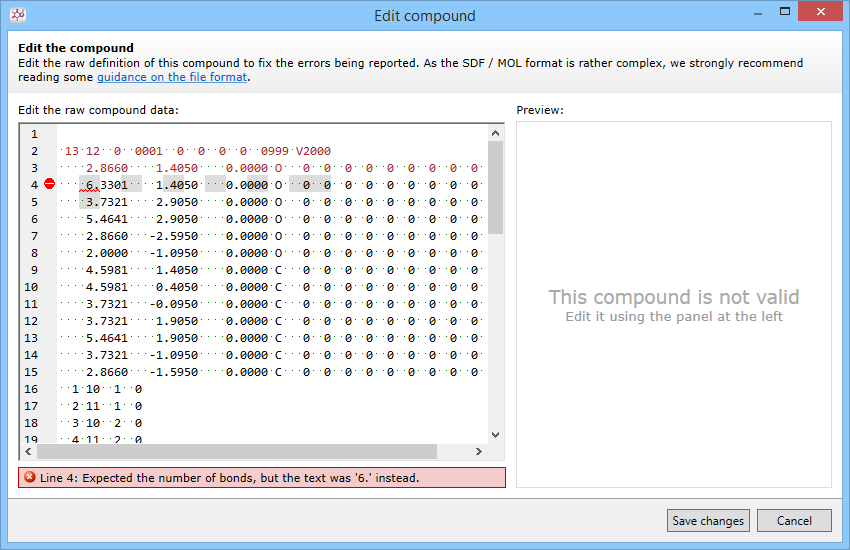
Compounds begin with three lines - name, program information, and a comment. Though these lines are permitted to be blank, there must be at least a line there. Often this can be a problem when sharing compounds over email. To fix this problem:
- Make sure you copy or paste the entire record (including those blank lines)
- Share compounds as attachments rather than in the body of the email, so you get the entire file
- Insert the necessary lines before the counts line using the Edit this compound tool in Progenesis SDF Studio.
Collapsed spaces
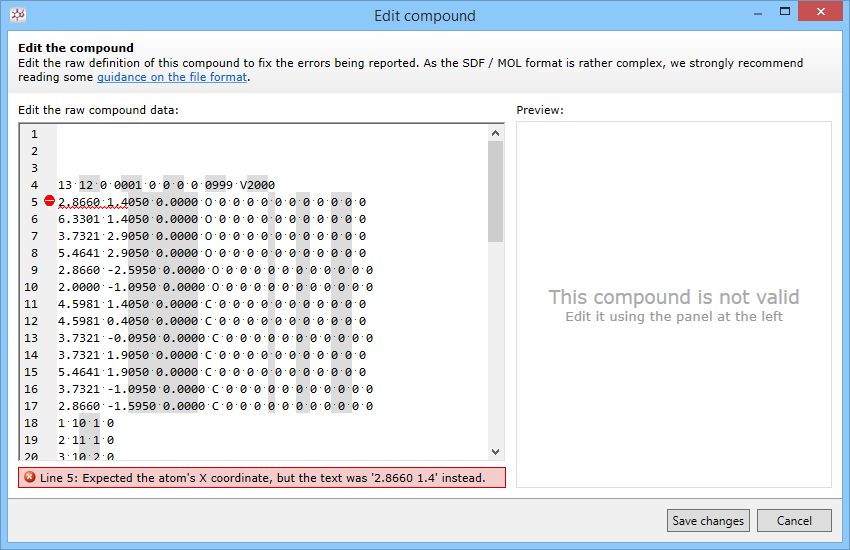
Compound information is recorded in a series of fixed length lines. Often, when sharing compounds in online forums or newsgroups, your browser will skip the extra spaces between the columns. To fix this problem:
- Use features of the forum software to share compounds as a file attachment or as a "code snippet"
- Add appropriate numbers of spaces to the counts line as well as all of the lines in the atoms, bonds, and properties block using the Edit this compound tool in Progenesis SDF Studio.