




What's new in the latest version?
The new features available in version 3.0 are summarised below. For more detail, you can download the update flyer (670KB).
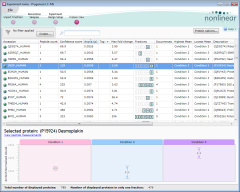 A new workflow to support analysis of fractionated samples
A new workflow to support analysis of fractionated samples
Fractionated samples can now be analysed and recombined to give a clear, and statistically robust, view of overall protein behaviour in the samples. The new fractionation workflow workflow is a very simple, yet very powerful, addition to Progenesis LC‑MS.
Resolve peptide conflicts faster and more reproducibly
Any features with conflicting peptide identifications will be excluded from the measurements for the affected proteins. Also, where a protein's set of identified peptides is a subset of another protein's peptides (e.g. as is the case with subunits), both will be grouped as being part of the protein with greater sequence coverage.
Both of these options are standard for any new experiments, and can be enabled for older experiments.
Ability to normalise feature abundances to a spike of known concentration
If you're using a spiked sample, or otherwise have a sample in which a signficant number of the proteins are affected by experimental conditions, you now have the flexibility to normalise to a subset of features.
Direct import of Scaffold peptide identification results
Ease the process of peptide conflict resolution by using Scaffold's percentile scores for peptides to inform your decision on which identifications to reject.
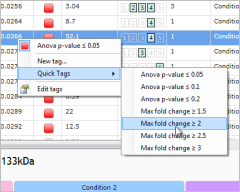 More powerful searching and filtering
More powerful searching and filtering
After gathering extensive usability feedback, we've improved the way in which you can find and filter the peptides of experimental interest. As well as being much simpler to use, the new tagging interface also has the power to create more complex filters with just a few clicks. You can read more about the new features in the tagging FAQ.
Additional improvements
- Advanced searching and filtering using regular expressions
- Improved contextual help, including videos
- Mass error column for peptide identifications
Read about all the features in our unique analysis approach.