




On this page
Advantages of label-free LC‑MS for quantitative proteomics
Labelling approaches, including the use of spiked synthetic peptide standards, are established for reliable quantitative studies. But when we talk with people in proteomics labs, there's a growing interest in applying label-free LC‑MS approaches. Especially with articles[1] from the likes of Matthias Mann (Max Planck Institute for Biochemistry, Martinsried, Germany), and other publications we've highlighted below, showing the major advantages that label-free LC‑MS gives you.
For example, there's evidence that label-free methods provide higher dynamic range of quantification[2,3]. This means you can measure significant changes within a complex mixture or across an entire proteome in a single experiment. Although it's worth noting that for spectral counting, this comes at the cost of unclear linearity and relatively poor accuracy.
The other key advantages of label-free LC‑MS approaches, including the differences between ion abundance based measurements compared to spectral counting can be summarised by tables taken from two publications[2,3].
| Application | Accuracy (process) | Quantitative proteome coverage | Linear dynamic rangea | |
|---|---|---|---|---|
| a In MRM mode, dynamic range may be extended to 4-5 logs | ||||
| Metabolic protein labelling |
|
+++ | ++ | 1-2 logs |
| Chemical protein labelling (MS) |
|
+++ | ++ | 1-2 logs |
| Chemical peptide labelling (MS) |
|
++ | ++ | 2 logs |
| Chemical peptide labelling (MS/MS) |
|
++ | ++ | 2 logs |
| Enzymatic labelling (MS) |
|
++ | ++ | 1-2 logs |
| Spiked peptides |
|
++ | + | 2 logs |
| Label-free (ion abundance) |
|
+ | +++ | 2-3 logs |
| Label-free (spectral counting) |
|
+ | +++ | 2-3 logs |
| iTRAQ | Label-free | |
|---|---|---|
| Protein loading | 800ug total loading (100ug per iTRAQ labelling vial) | 0.5ug for each of 3 technical replicates |
| Number of overnight steps | 5 | 1 |
| Samples to analyse by MS | 30-60 fractions | 1 per condition |
| RP-LC and MS acquisition | 30-60 hours | 2 hours |
| Total analysis time | 6 days | Less than 3 days |
| Total instrument time | 30-60 hours | 6 hours/sample |
| Size of data file | 300MB × 40 | 6GB × 3 |
| No of proteins confidently identified (>1 peptide) | 178 | 421 |
| Average no of peptides per protein (including single peptide id's) | 5 | 12 |
| Average sequence coverage | 11% | 45% |
Evidence of label-free approaches working for our customers
We worked with researchers from the University of Conway Dublin using label free LC‑MS to analyse proteomic changes in rat livers with significant liver damage. The results were presented as a poster during the 2009 BSPR meeting in Cambridge[4]. They showed:
- Excellent analytical reproducibility, with average CV = 10.9%
- Good proteome coverage (595 proteins identified at 6% false identification rate)
- Modest instrument time required per sample
- The data produced could be used to help perform pathway analysis
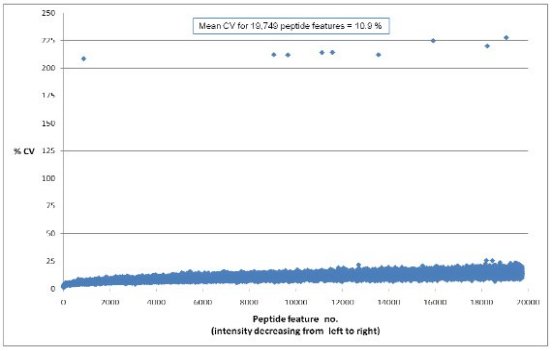
Figure 1: Plot displaying the % coefficient of variation across 6
technical replicate injections of a pooled liver sample. Virtually all of the peptides are below a
CV of 25% and the mean is 10.9%[4]
Progenesis LC‑MS has simplified and enhanced the analysis of label free LC‑MS data at the Conway Institute....The inclusion of embedded statistics functions and simple data reporting and export make integrating label free LC‑MS datasets into established workflows considerably more straightforward than has been previously possible.
Professor Steve Pennington, UCD Conway Institute, Ireland and Vice and President of The British Society of Proteome Research (BSPR)
Benefits of new approaches in data analysis software
Despite the advantages of label-free, it's been a struggle for everyone to realise them. Without suitable software, analysis of this data is challenging, particularly if your quantification approach relies on detection and identification of known, unique peptides for each protein of interest. Complex data analysis must be simple to use and easily integrate into your existing workflows, if it's going to make an impact in your proteomics research.
Progenesis QI for proteomics, however, is being adopted for label-free LC‑MS data analysis by some of the leading research groups. We hope you'll be able to share the benefits they've been enjoying.
Sources
- Comparative analysis to guide quality improvements in proteomics, Matthias Mann. Methods 6 (Oct 2009), pages 717 - 719
- A Comparison of Labelling and Label-Free Mass Spectrometry-Based Proteomics Approaches. Vibhuti J. Patel, Konstantinos Thalassinos, Susan E. Slade, Joanne B. Connolly, Andrew Crombie, J. Colin Murrell and James H. Scrivens. J. Proteome Res., 2009, 8 (7), pp 3752–3759. May 12, 2009
- Quantitative mass spectrometry in proteomics: a critical review. Marcus Bantscheff, Markus Schirle, Gavain Sweetman, Jens Rick, Berhard Kuster Anal Bioanal Chem (2007), 389:1017-1031
- Label Free LC‑MS Based Proteomics for Integrated Preclinical Pharmaceutical Toxicology, experience from the FP6 InnoMed PredTox Consortium (918KB) Ben C. Collins, Martin Wells, William M. Gallagher, Stephen R. Pennington. Scientific poster produced by researchers at the University College Dublin, Ireland for the BSPR meeting, Summer 2009.