




How can I perform a spectral library search?
Progenesis can search spectral libraries in .msp or .sptxt formats. To perform a search, first you need to download or create a spectral library.
Where can I find public spectral libraries?
Progenesis can support most .msp and .sptxt files, as long as they contain the required information.
Some public spectral libraries can be found at the following locations:
- NIST Libraries of Peptide Tandem Mass Spectra (.msp)
- SWATHAtlas Libraries (.sptxt)
- Spectral libraries created from Mascot (.msp)
How do I create my own spectral library?
You can create a spectral library from your own experiments using the following steps:
- Perform an analysis and search for identifications using one of the peptide search engines. (Note: if you are performing a search of Waters MSᴱ, HDMSᴱ or SONAR data using a search engine other than Ion Accounting, you must provide the exported MGF file when importing your results).
- Use the Refine Identifications and Resolve conflicts screens to filter identifications to only those you are confident are correct. This is an important step as Progenesis will export all possible identifications that haven't been deleted or rejected.
- If desired, apply a peptide ion tag filter to show only the ions whose identifications you wish to export to the spectral library.
-
At the Identify Peptides screen, choose Export spectral library… from the File menu.
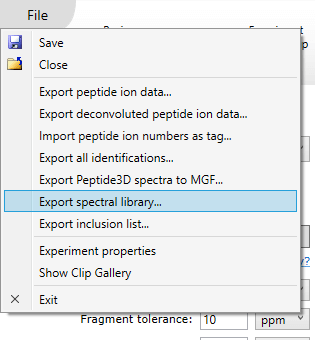
- Choose a file to export your library to.
- If exporting to an existing spectral library, you can choose to Overwrite or Append to the existing library.
How do I perform a spectral library search?
-
At the Identify Peptides screen, choose Spectral Library Search in the identification method dropdown. This will show you the following interface:
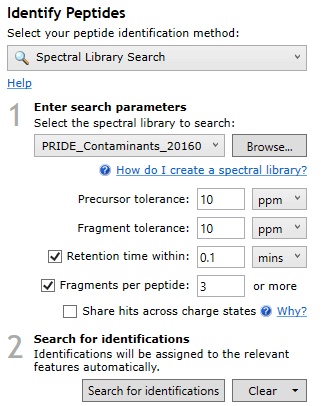
- Choose or browse to the spectral library file (.msp or .sptxt) that you would like to search.
- Enter your desired search parameters (see below for more information).
- Click the Search for identifications button.
Spectral library search parameters
- Precursor tolerance
- Specifies the mass error below which a precursor is considered a match to a spectral library entry.
- Fragment tolerance
- Specifies the m/z error below which a fragment is considered a match to a spectral library entry's fragments.
- Retention time within
- Specifies the retention time error, below which a precursor is considered a match to a spectral library entry. If this is not selected, or a spectral library entry has no retention time information, it has no effect.
- Fragments per peptide
- The minimum number of fragment matches required for a match to be accepted. If not selected, all matches are accepted, even those with no fragment matches. If a spectral library entry has fewer fragments than the given value, all of its fragments must match. If a spectral library entry has no fragments, matches to that entry will be accepted regardless of this value.
- Share hits across charge states
- If selected, charge state deconvolution of identifications will be performed. This means that if only some charge states of a given peptide were identified by the spectral library search, the other charge states will inherit the same identifications.
Sharing spectral library search hits across charge states of peptides
When identifying peptides in Progenesis, the ions in your experiment may inherit identifications from other ions considered to be different charge states of the same peptide. This is to help with accurate peptide-level quantitation in the absence of complete search results. In the Spectral Library search, however, this behaviour can be overridden.
If you want to avoid a particular charge state of a peptide being included in its protein's quantitation — e.g. due to it co-eluting with an ion whose MS1 signal interferes with that charge state's signal — you may choose to not export spectra for that charge state to your spectral library. In subsequent experiments, to avoid that ion's identification being inherited again during a spectral library search, clear the Share hits across charge states option. Identifications won't get assigned to peptide ions excluded when the spectral library was built.
Bear in mind, however, that this is a trade-off. You may lose the benefits of greater quantitation accuracy in some proteins while gaining the benefit of less re-review and exclusion of ions for other proteins. If you're unsure of which option is best for you, we recommend you leave the checkbox selected to gain the quantitation benefits.
What must my spectral library contain?
Most public spectral libraries in .msp or .sptxt format, along with those created by Mascot or Progenesis QI for proteomics will be searchable by Progenesis.
However, for completeness we list here the information required by Progenesis to perform a spectral library search. A minimal MSP entry would look like this:
Name: QADREGYPEVAEAYK/2 Comment: Parent=863.4075 Protein="gi|126699128 rubrerythrin [Clostridium difficile 630]" Mods=0 Num peaks: 0
The following example shows an entry that contains fragment information and the optional fields that Progenesis understands:
Name: QADREGYPEVAEAYK/2 Comment: Parent=903.3907 Protein="gi|126699128 rubrerythrin [Clostridium difficile 630]" Mods=1/6,Y,Phospho RetentionTimeMins: 21.288 ProteinMass: 20319.000 Start: 20 End: 34 Num peaks: 2 706.4592 277.2237 803.2977 170.8182
If any of the entries in your spectral library don't contain the necessary information, you will see the following warning:
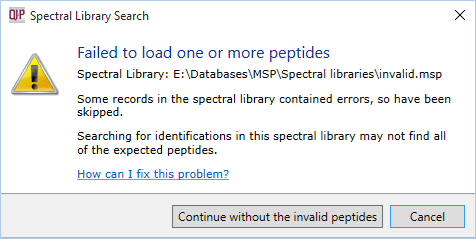
You can choose to Continue without the invalid peptides if you wish. This may result in fewer identifications than you expect, as invalid entries will be ignored.