




Updating from Progenesis LC-MS v2.0 to v4.1
Automated data processing
Reference run selection and alignment can be started, and the runs will be processed automatically as they load . You can leave it to process without any further interaction.
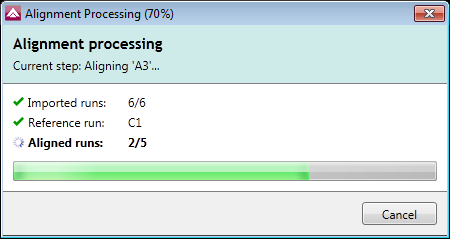
Automatic alignment reference selection
The alignment reference can be selected automatically by the software, increasing the objectivity and reproducibility of your analysis results.
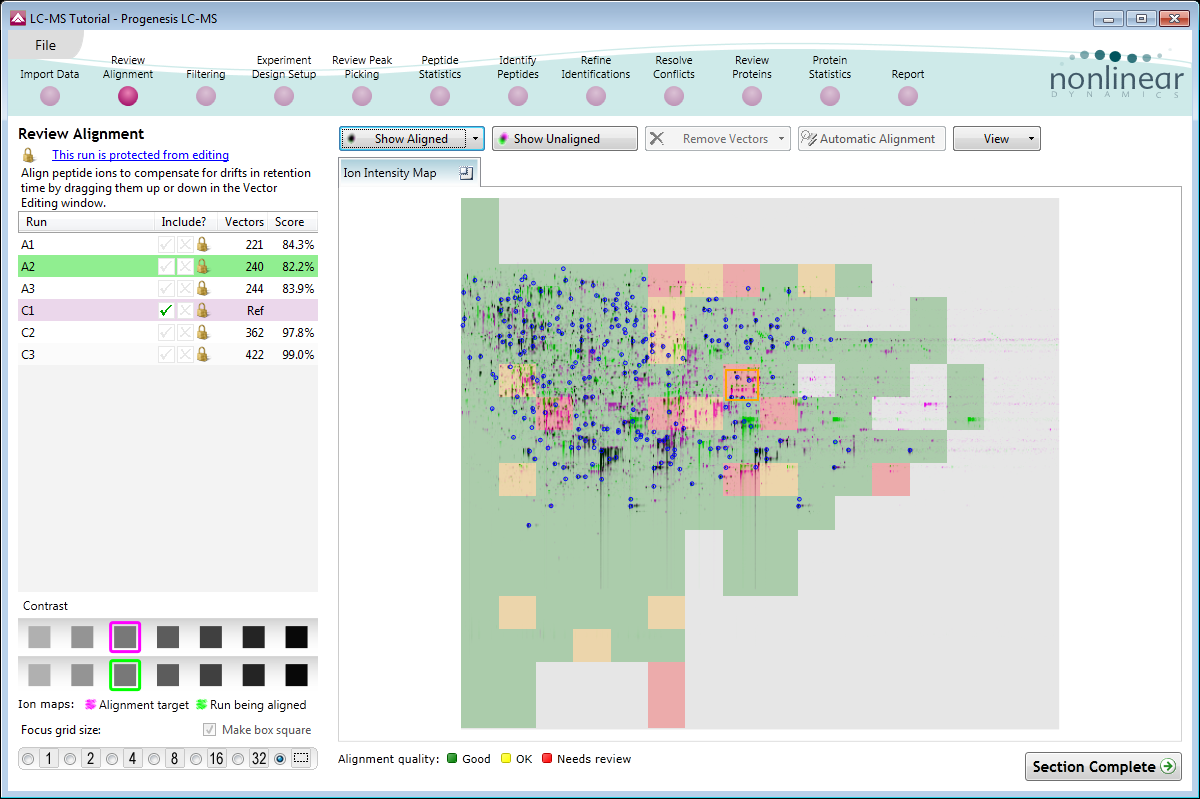
Alignment QA measures and visual display
You can review the quality of automatic alignment, giving you confidence that downstream analysis steps will deliver the most reliable results.
Experiment design set-up from file
Your experiment design can be set up from an imported file which contains your sample groupings.
Fast review of any step in a completed analysis
You no longer have to move stepwise through all of the analysis steps when reviewing your analysis.
Clip gallery
Capture print quality images from some of the most important data displays, which can be used in posters and presentations.
Progenesis improvement program
This opt in program provides information on software usability, amongst other things, to our software engineers which can help direct future development.
Extended workflow for the quantification and identification of proteins
Automatically generate a tabular and graphical view for all of the proteins of interest that you determine within your experiment.
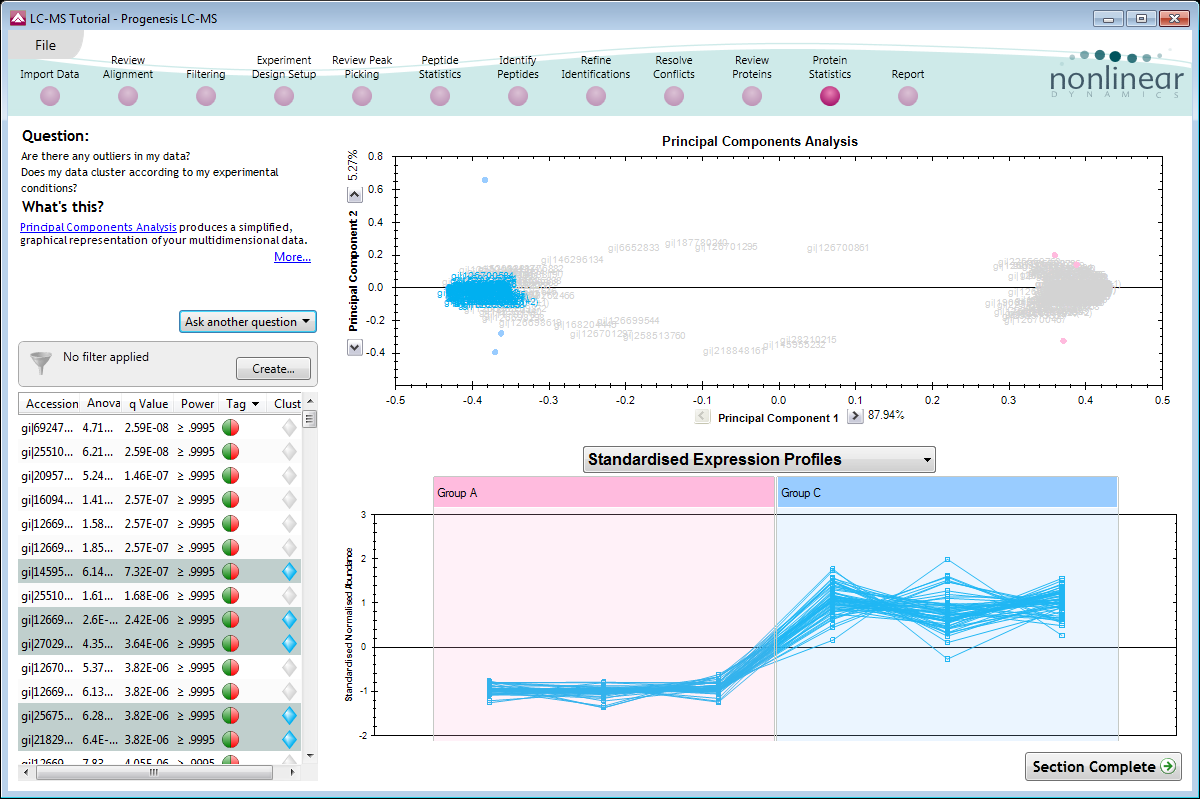
Multivariate statistics on protein expression data
Includes q-value, to control false discovery rates, principal components analysis (PCA), correlation analysis and power analysis.
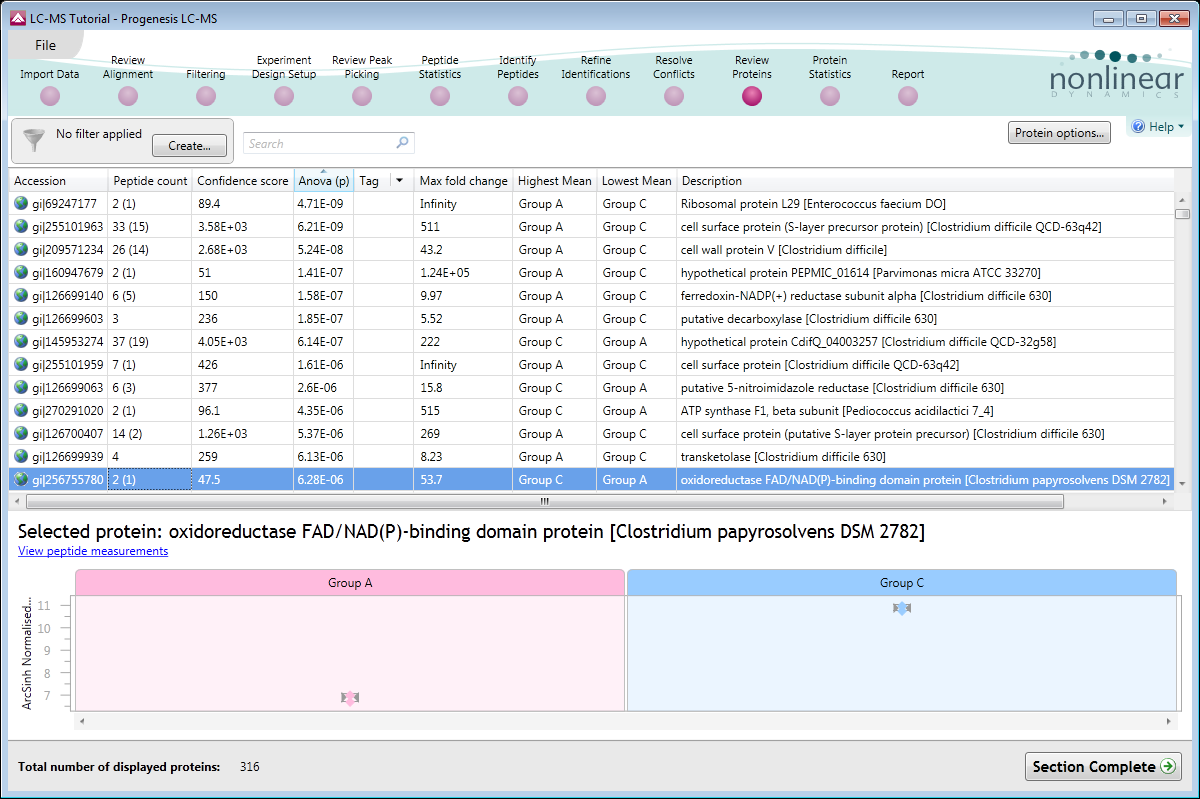
Protein reports
Report a protein (top-down) as well as a peptide (bottom-up) view of your proteomics experiment.
Gas-phase fractionation
Support for increasing protein and proteome coverage using a gas-phase fractionation approach.
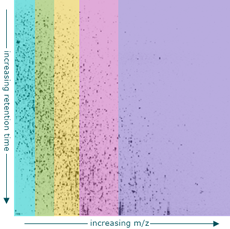
Data formats
Supports direct loading of Agilent .d data as well as the mzML file format.
Fractionated sample analysis
Perform label-free quantification and protein identification to each fraction separately. The fractionation workflow then combines all of these fractions into a single protein based view of the experiment.
Update to normalisation methods
You can normalise to all proteins, a set of housekeeping proteins, or opt not to perform any normalisation.
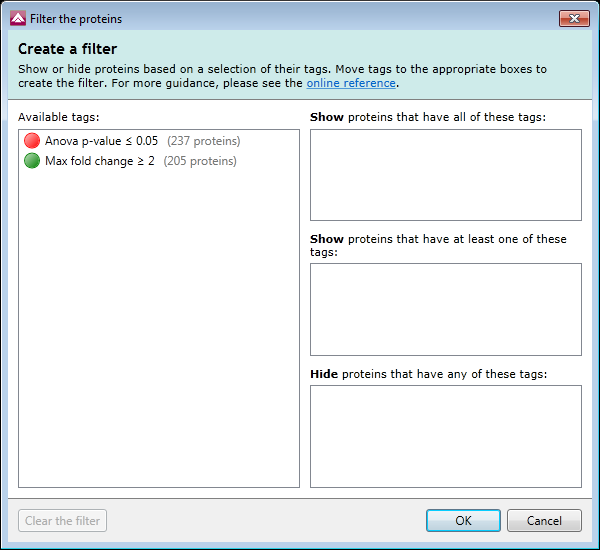
Improvements to tags and searches
There is a new filter dialog which makes it easier to reduce the data you are viewing so you can focus on the real peptide ions of interest.
Link with scaffold
Import Scaffold results into the software.
Full control over detection parameters
You can adjust the sensitivity of the detection algorithm for peak picking, including the option to set a minimum retention time window and select which runs you wish to use for peak picking.
Improvements to peak picking
Less fragmentation in the time dimension and noticeable improvements in the algorithm’s ability to distinguish between streaks and real peptide ions.
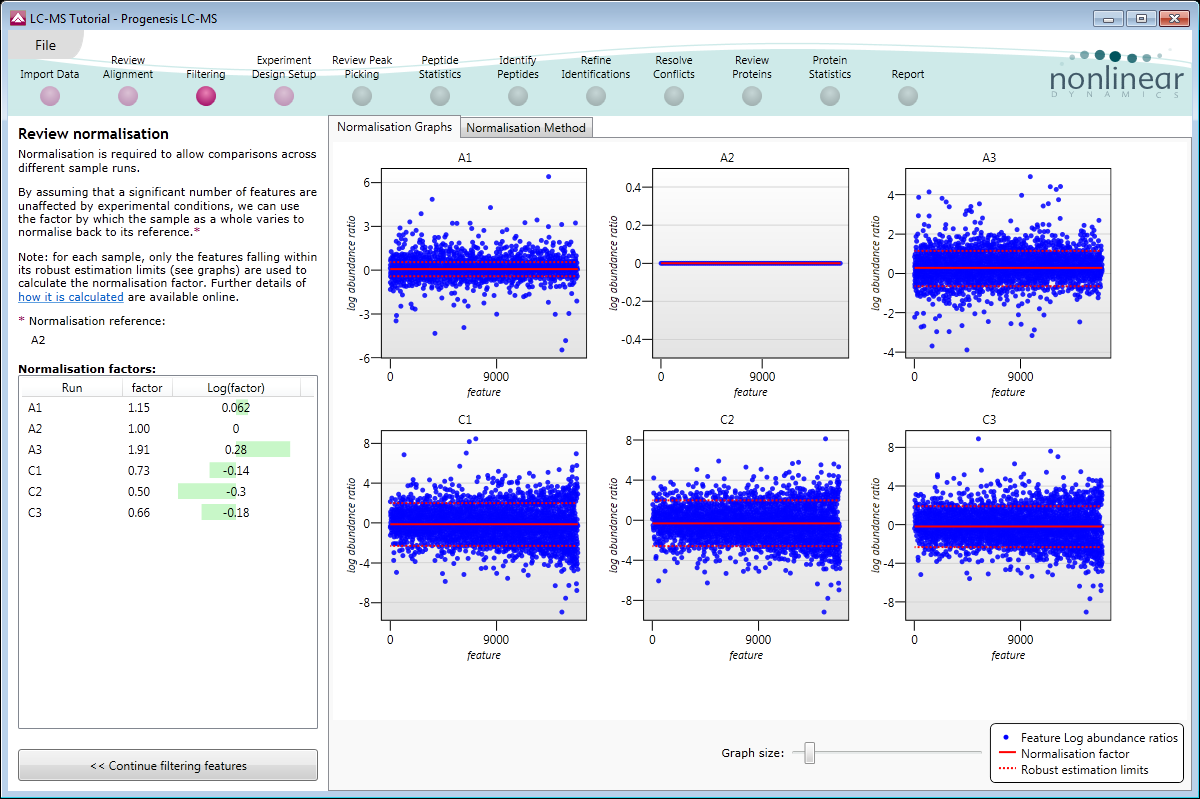
Visualisation of normalisation
Normalisation is visualised on a scatter plot and a normalisation factor is calculated for each run so you can assess the results of normalisation on your experiment.
Calculation of Coefficient of Variation (CV)
CV measures help you determine the variation within an experimental condition.
Addition of a second experiment design option
"Within Subject Design" allows the comparison of different samples from the same subjects under different conditions. An example of this would be a time course experiment.
Improved compatibility with Waters' instruments
The software can handle lock mass correction for .RAW format data and import peptide identification results from Waters' ProteinLynx Global Server (PLGS).
Total ion chromatogram (TIC) view
Aids alignment verification providing an additional quality check.
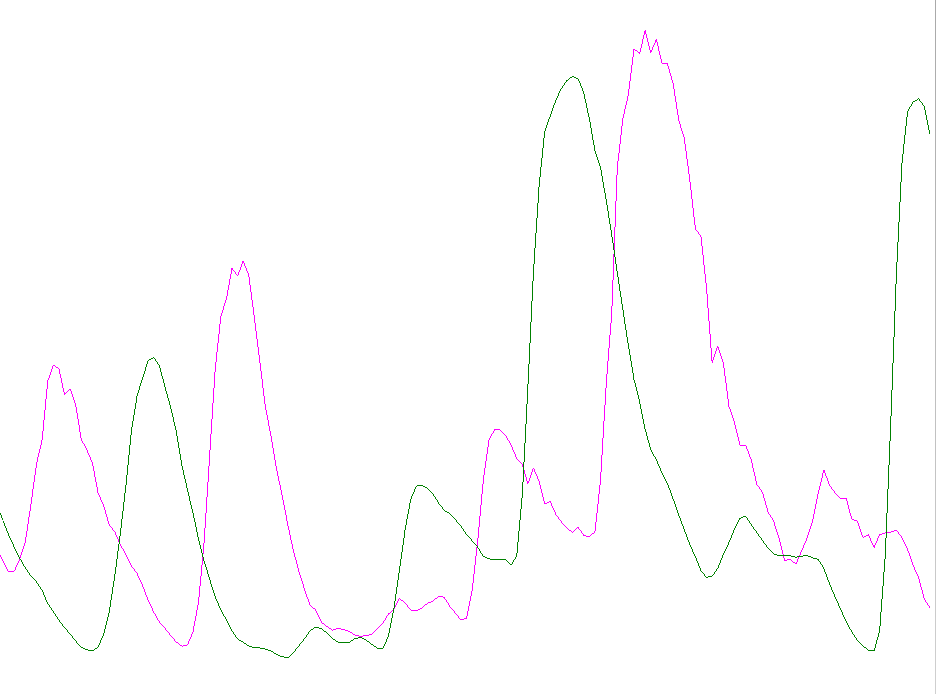
Before alignment

After alignment
Manually added alignment vectors override automatically generated vectors
Increasing the weighting on manual vectors gives you more control over the alignment process and makes it easier to accurately align difficult data.
Filtering of MS/MS spectra prior to export
Enables the removal of spectra from your search before they are exported for identification, reducing the amount of data sent to the search engine.
MS/MS peak processing
Reduces the data file size by removing the noisy peaks, retaining only the essential data required by the peptide search engines. Also improves the quality of search results obtained.
Peptide filter step
Filter out identification results, rapidly reducing the number of incorrect assignments, so you can save time reviewing search results.