



Support for ChemSpider
Support for this compound search method is provided as standard.
About this plug-in
ChemSpider is a web-based chemical structure database with access to over 32 million structures from hundreds of data sources. This method makes use of those ChemSpider web services, automatically exporting data from Progenesis QI to ChemSpider for searching according to the parameters you select, importing the results, and assigning them against the correct compounds within the software. This service is intended for users with a valid support contract, or users who are evaluating, and may be restricted to those users in the future.
Selecting this search method
At the Identify Compounds step of the workflow, use the drop-down menu to select ChemSpider:
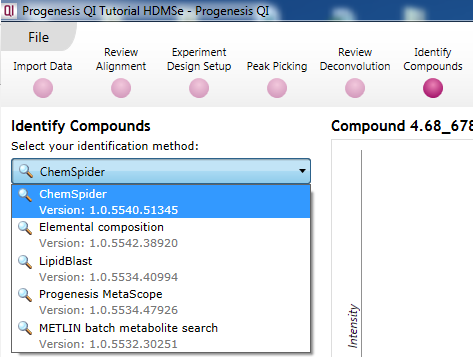
The drop-down selection dialog at Identify Compounds.
Filter the compounds
You may wish to select a filtered set of compounds for this process, to reduce the processing and curation time. This is because ChemSpider can search a very large range of data and return a large number of results depending upon the search settings. This can be done using tag filtering. For example, you may wish to only search compounds that are significantly altered between control and treatment groups. The search will be carried out for all compounds present in the current filter (or all compounds in the experiment, if no filter is applied).
Set the search parameters
The ChemSpider method requires just two selections to be made; the mass error tolerance and the data sources to be searched.
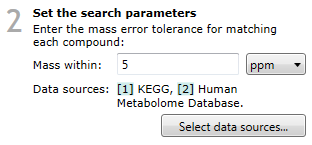
Search parameter selection.
The mass error tolerance, in ppm or Da, is the maximum difference between the mass for the compound in the databases searched and the observed mass. Where a compound has been successfully deconvoluted, the observed neutral mass is searched directly against the database neutral mass.
Where only a single m/z is available, and its adduct status is unclear, a single neutral mass cannot be assigned for searching against the database(s). Instead, this compound will be searched multiple times. Each time, it is treated as if it is derived from a different adduct; the neutral mass corresponding to that original m/z and assumed adduct is calculated and searched, and this is repeated for all the adducts you defined in the experiment setup that could impart the correct observed charge state for the compound. This allows attempted identification even for compounds without a known neutral mass.
The second parameter is the selection of one or more data sources from the available list. On clicking the Select data sources button (as seen in the image above), you will see the following dialog:
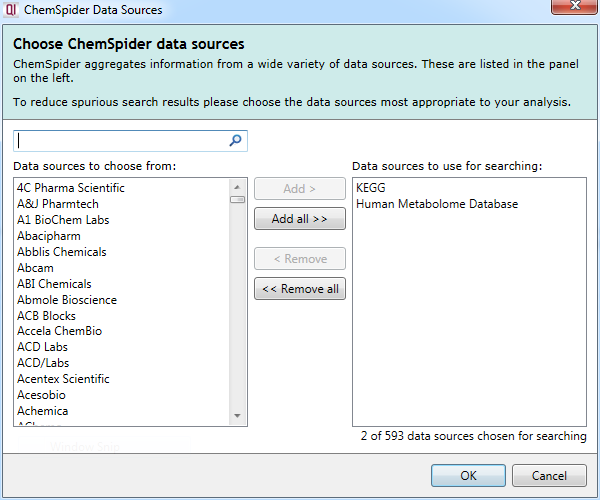
The ChemSpider Data Sources selection box that appears on clicking on the Select data sources button.
You can select or deselect data sources individually, use multiple selections (e.g. Shft- or Ctrl-clicking), select or deselect all data sources at once, and can track down data sources of interest using the Search box. Once selected, the data sources are listed on the main page in the search parameter selection section, as was shown earlier (e.g. KEGG, Human Metabolome Database). Up to the five will be listed by name, and any after that presented as "...and X more".
Search for identifications
Once your parameters are set up, clicking on this button will begin the search process.
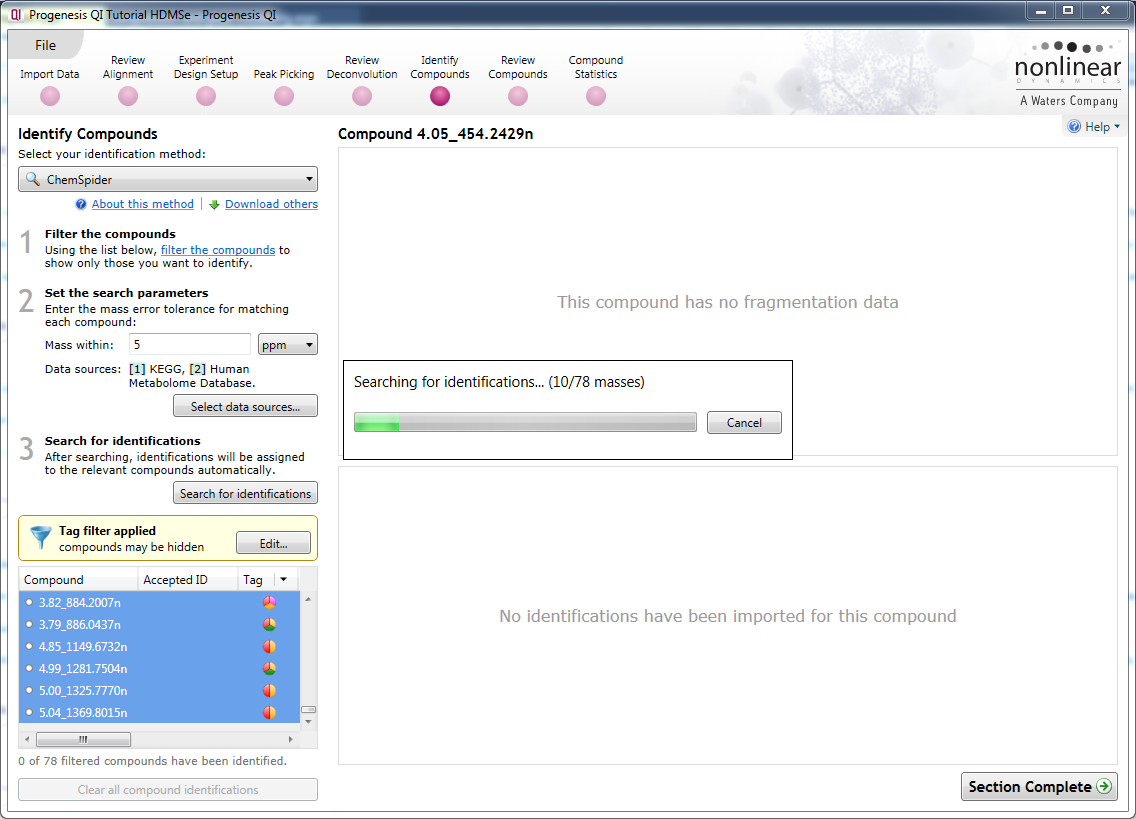
A ChemSpider search in progress.
Minimising search time
Given the number of databases (potentially) searched, ChemSpider searches can take a long time to complete. To minimise this, you can pre-filter your compounds, as described above. You may also wish to narrow your database selection only to highly relevant databases rather than including a larger number, and narrow your mass error tolerance as much as is reasonably possible for your instrument. These measures will reduce the search space, and return results more rapidly.
Results
Identifications are obtained, then imported and matched against the relevant compounds automatically. The hits from a given compound will be denoted by ChemSpider IDs (CSIDs); these values will return more information if entered as search terms into ChemSpider, including the data sources for the compound. To save manually entering the data, the Link field of the table also provides a direct path to the relevant page at ChemSpider.
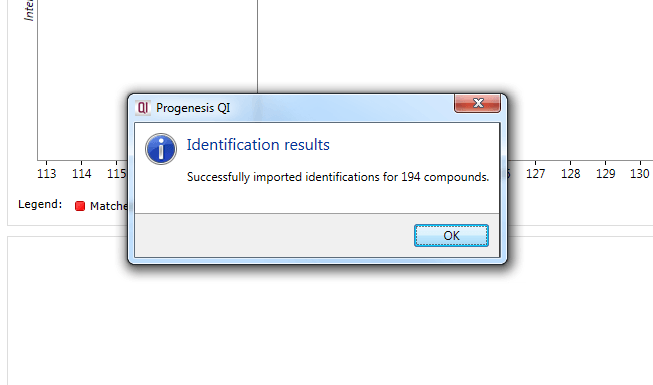
The notification obtained upon search completion.
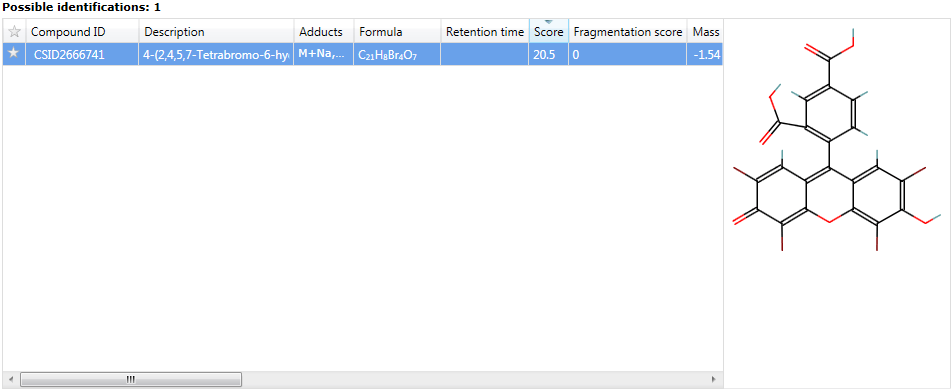
Identifications are automatically added to the Possible identifications table.
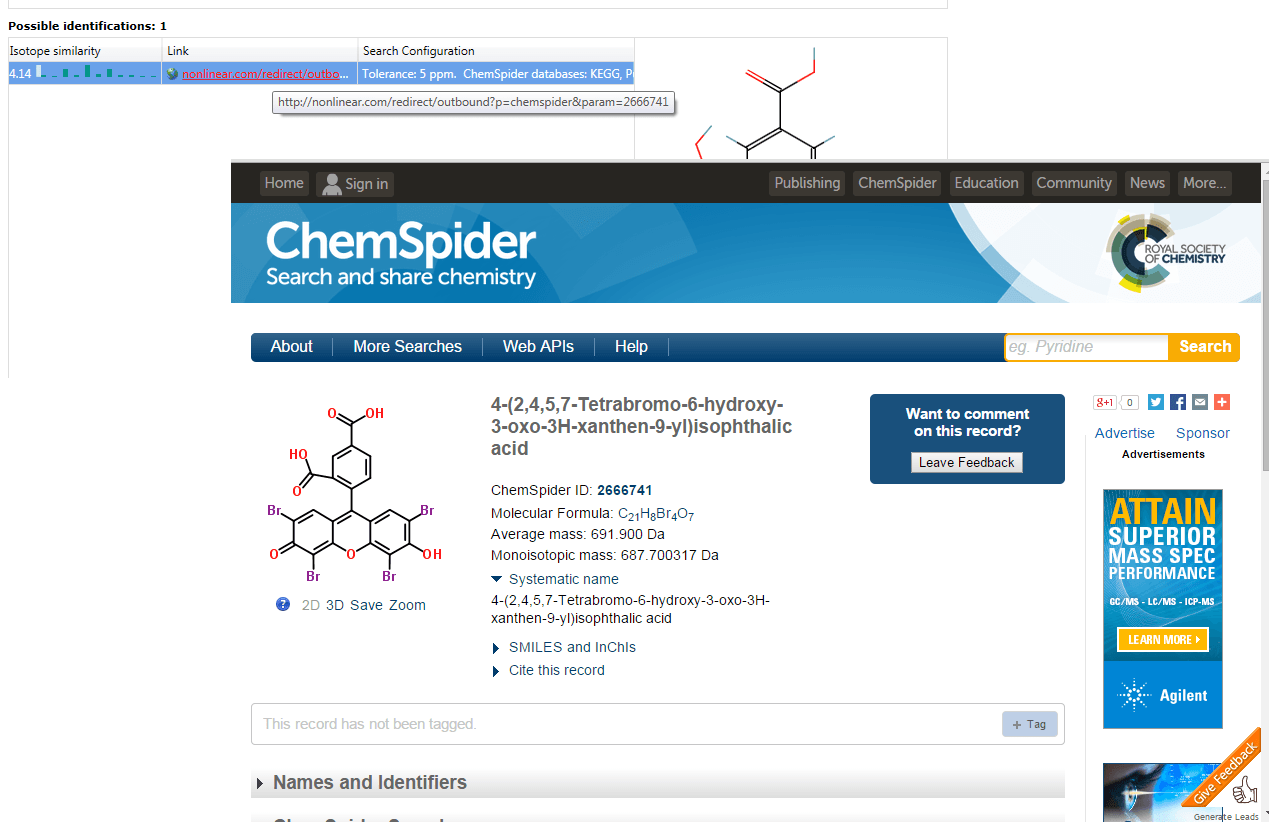
Clicking on the entry in the Link field will redirect you to the ChemSpider entry for a hit.
Multiple searches
Note that if you perform one ChemSpider search but select multiple databases, this is imported as one search by the software. The external database that the identity was discovered in is not marked, as the CSID is a unique identifier across all of ChemSpider. To determine where a ChemSpider hit resulted from in this case, the CSID or link should be used to query ChemSpider directly, where the data sources containing that CSID are displayed.
If you perform two separate ChemSpider searches, with different parameters, and obtain new identification data, then results from the two searches will be assigned a unique letter by the search they came from, to differentiate which results came from which search.
When letters are assigned, a compound that would have been identified by more than one of the searches would be given the letter of the first search that discovered it (i.e. a compound returned by both searches A and B carried out in that order would be assigned the identifier for search A).