



Support for Elemental composition
Support for this compound search method is provided as standard.
About this plug-in
The elemental composition method calculates theoretical molecular formulae that match the measured mass and isotopic distribution of the experimental compounds. Each result is a list of possible atomic species present in one compound molecule (e.g. C6H12O6).
Using the method
To use this method, select Elemental composition from the drop-down menu at the Identify Compounds step of the workflow:
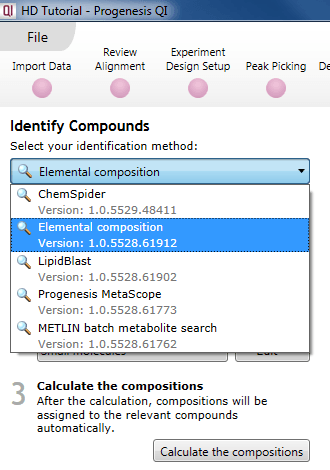
The Elemental composition selection dialog.
Filter the compounds
You may wish to select a filtered set of compounds for this process, to reduce the processing time. This can be done using tag filtering. For example, you may wish to only calculate molecular formulae for compounds that are significantly altered between control and treatment groups. The calculation will be carried out for all compounds present in the current filter (or all compounds in the experiment, if no filter is applied).
Set the calculation parameters
Three default parameter sets are provided, with suggested settings to suit certain classes of analytes. You can use one of these default sets to carry out the calculations, customise them, or create your own.
Using a predefined set
The default parameter sets are "Small molecules", "Lipids", and "CHNO" (which is optimized for simple organic compounds). These can easily be selected using the drop-down menu.
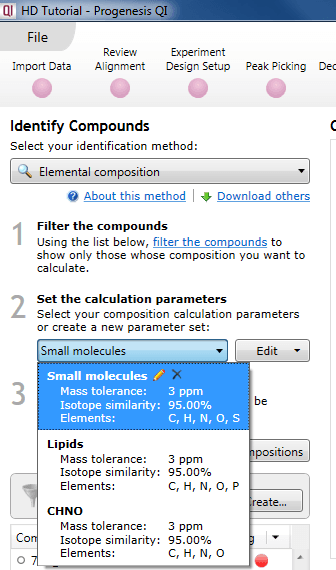
Selecting an existing parameter set.
Setting your own parameters (optional)
Should you wish to make your own set of search parameters, you can either edit a pre-existing one, create a copy of a pre-existing one and edit that, or make an entirely new set. This can also be initiated via a simple drop-down menu.
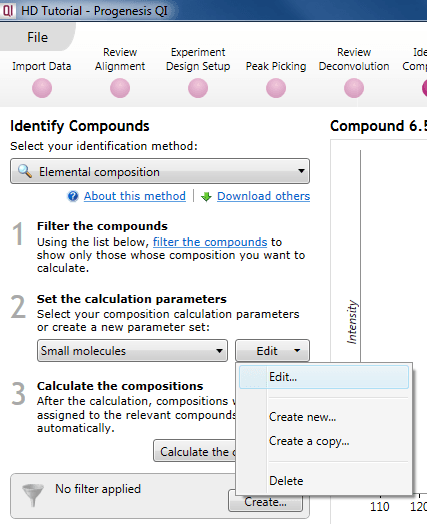
The dialog for editing or creation of a new set.
In all cases, the interface is the same, based on a periodic table:
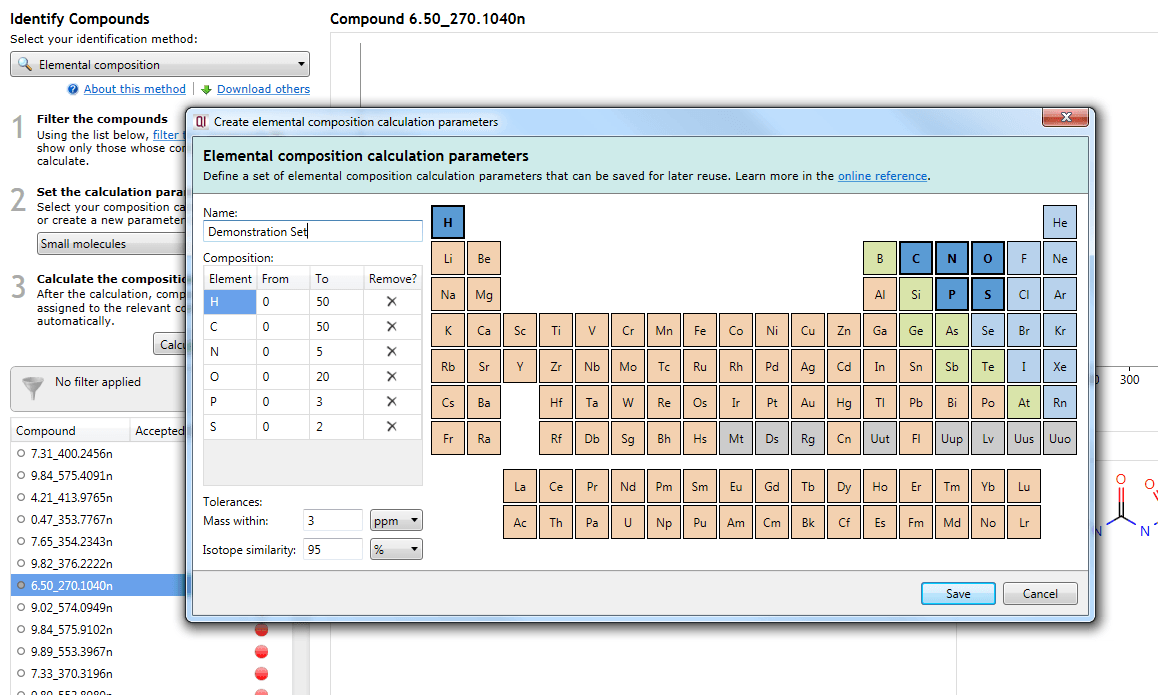
The interface used to define or edit a set.
Adding/removing an element and setting its ranges
Clicking on an element in the table will add it to the calculation, allowing it to be considered as part of the possible molecular formulae. The element will then be listed in the Composition table below the parameter set name.
The From and To fields are customisable and allow you to specify the minimum and maximum number of atoms of each element (range) in calculated molecular formulae.
To remove an element from consideration, you can press the Delete key with that element selected in the Composition table, click on the cross button in the Remove? column of that table, or click on the element in the periodic table a second time.
Note that the more elements there are in your parameter set, and the wider their ranges, the larger the possibility space is, and the longer the calculation will take. If you find that the calculation is taking too long to complete, you could try reducing the numbers and/or ranges of elements. You may also find that narrowing these parameter sets will return fewer possible results for further investigation, aiding your assignment of identities.
Also, higher mass compounds will take longer to search than lower mass ones, because of the greater number of possible combinations to consider in the calculation.
Setting the mass error
The mass error is the allowable difference between the actual mass of your compound as determined by the software, and the mass of the calculated molecular formulae (in Da or ppm).
Note that as this threshold is raised, the processing time and number of potential results will rise.
It should also be noted that molecular formulae can be calculated both from a deconvoluted neutral mass, and where only an m/z value is available such that the compound cannot be deconvoluted to a single neutral mass. In the latter case, possible formulae will be calculated assuming that the m/z originated from each adduct in the experiment that could impart the correct charge state to the compound, with the implied neutral masses for each case used in the calculation. This mass error setting applies in all such cases.
Setting the isotope similarity cut-off
The isotope similarity cut-off can be set by minimum percentage. This is calculated as a similarity score between the theoretical and observed isotope patterns in the same manner as for the Metascope scoring engine.
Carrying out the calculation
After setting and saving the parameters, you can select Calculate the compositions to proceed.
Results
The results of an elemental composition search will be assigned against compounds in the Possible identifications table at the Identify Compounds step of the workflow; the results will be assigned as a row to the compound identifications and given a new letter if previous searches have already been carried out. In this row, the text "Formula XYZ determined by elemental composition" will be displayed in the Description field, and the calculated formula presented in the Compound ID and Formula field. A URL will be provided for every calculation in the Link field of the table; this will automatically display the results of searching the calculated molecular formula using the NIH Chemical Structure Lookup Service (CSLS).
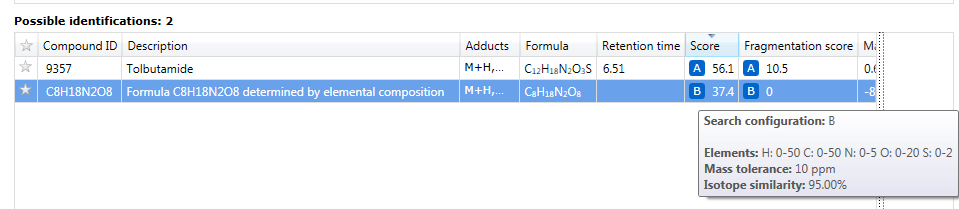
Elemental composition results added to a compound.

A link out to the CSLS result for a compound.
How elemental composition can help with identification
Theoretical molecular formulae may provide clues as to the identity of a metabolite where direct database matches are not forthcoming. The calculated formulae may be manually searched in online databases such as Chemspider, PubChem and the NIH CSLS, for example, to return potential identifications. As previously, the latter can be opened by default by clicking on the link in the results for the compound; others can be searched manually.