



Support for LipidBlast
Support for this compound search method is provided as standard.
About this plug-in
LipidBlast is a computer-generated MS/MS database, produced by the Metabolomics Fiehn Lab [1].
Lipid fragmentation MS/MS data are made available in the form of two .msp files (one for positive ionisation mode and one for negative). The Progenesis QI LipidBlast search uses these .msp files, allowing you to search by both neutral mass and ms/ms.
Progenesis QI calculates a neutral mass from each lipid formula in the database, and matches the intact compound mass on this basis. Where a match is achieved, it then assesses the matching of observed fragments of that lipid to the MS/MS fragmentation pattern stored in the database.
Downloading and configuring the LipidBlast databases
The first time you select the LipidBlast search method at the Identify Compounds step of the workflow, Progenesis will ask you to download the LipidBlast .msp database files:
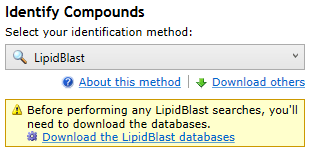
The prompt to download databases that appears on first selecting the LipidBlast search method.
This process is automatic once initiated. Once the databases have been downloaded and installed, the LipidBlast plugin will be configured for all future searches.
If you cannot access the internet from the specific computer running Progenesis, then you can transfer the files from another computer that does have internet access. Instructions for carrying out this step are provided separately.
Filter the compounds
You may wish to select a filtered set of compounds for this process, to reduce the time you spend curating the data, as LipidBlast may return a large number of results depending upon the search settings. This can be done using tag filtering. For example, you may wish to only search compounds that are significantly altered between control and treatment groups. The search will be carried out for all compounds present in the current filter (or all compounds, if no filter is applied).
Set the search parameters
LipidBlast searches require the selection of three search parameters:

The LipidBlast parameter selection options.
- Precursor tolerance
-
This value, in ppm or Da, is the maximum difference between the mass for the compound in the databases searched and the observed mass.
Where a compound has been successfully deconvoluted, the neutral mass is searched directly against the neutral mass calculated from the lipid formula.
Where only a single m/z is available, and its adduct status is unclear, a single neutral mass cannot be assigned for searching against the known lipid neutral mass. Instead, this compound will be searched multiple times. Each time, it is treated as if it is derived from a different adduct; the neutral mass corresponding to that original m/z and assumed adduct is calculated and searched, and this is repeated for all the adducts you defined in the experiment setup that could impart the correct observed charge state for the compound. This allows attempted identification even for compounds without a known neutral mass.
- Fragment tolerance
-
This threshold (in ppm or Da) is the maximum difference between the mass for a fragment of the compound in the databases and the observed mass of the fragment.
LipidBlast contains fragmentation data for all commonly adducted forms of lipids. Progenesis QI will search only for fragments that are derived from the adducted precursors it has determined are present for that compound, to reduce false positives (hits against fragments that would not be present as the precursor for them is not present).
Note also that where experimental fragmentation data are available for a compound (MS/MS was triggered on that compound peak, for example) then for a hit to be returned, there must be at least one successfully matched fragment as well as the neutral mass. Where there are no experimental fragmentation data, then a match only to the precursor will be acceptable to return a hit, as fragmentation evidence is not present to assess. However, compound fragmentation is reported in the Fragmented? column of the compound table, such that you can differentiate between these outcomes.
- Report isomers as a single search hit
-
When selected, the search will merge hits that differ only in double bond positioning and/or geometry (lipid sub-species with the same backbone and side chains, but with double-bond isomerism within chains).
For example, if a search returns two identifications:
- GPCho(16:0/20:3(8Z,11Z,14Z))
- GPCho(16:0/20:3(5Z,8Z,11Z))
these will be reported as a single hit, GPCho(16:0/20:3).
When this option is not selected, all lipid isomers will be returned as separate search hits. Read more about this option. However, in the majority of cases, LC-MS/MS analysis cannot distinguish between these isomers, so the grouping option is activated by default.
Search for identifications
Clicking this button will begin the search, and automatically import the results against your compounds at the Possible identifications window at the Identify Compounds screen.
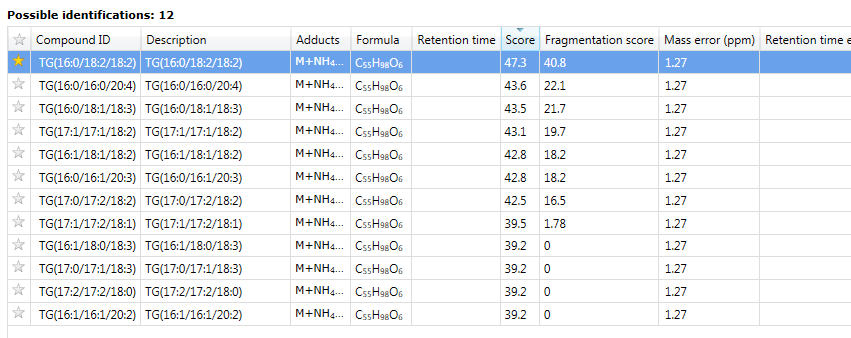
An imported LipidBlast search result, showing part of the Possible identifications table.
You can visualise the matching of measured to database-predicted fragments for each potential ID using the 'mirror plot' on the same screen, after clicking on a search result. Matched fragments are shown in red, unmatched in grey; and hovering the cursor over a fragment match will display relevant information in a tooltip.
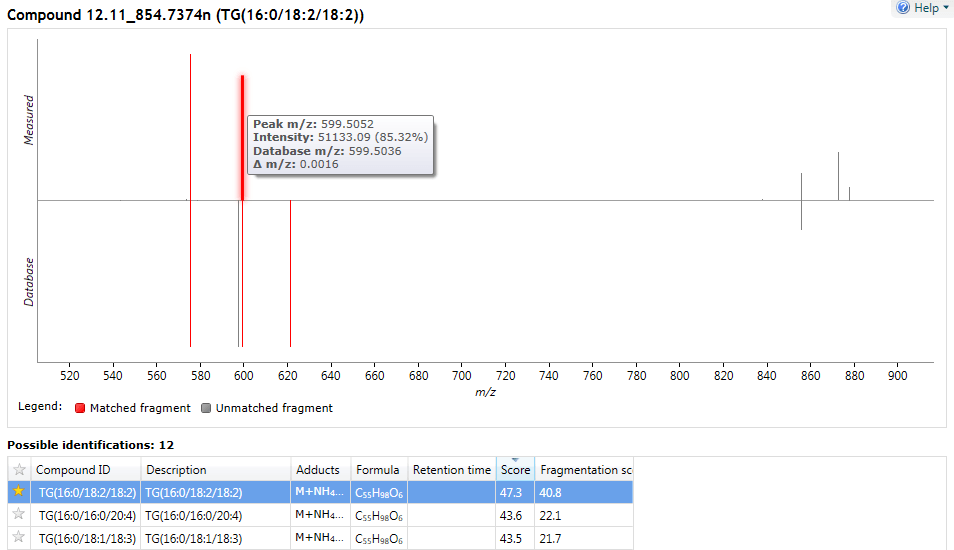
Fragment matching for LipidBlast results can be visualised in a mirror plot above the Possible identifications table.
Structure diagrams
LipidBlast itself does not contain structural data. If you would like to see the structural diagram for a LipidBlast result, click on the URL shown for the identification in the Link field of the Possible identifications table. This will take you to LIPID MAPS where you can see the structural diagram and further information about the identified compound.
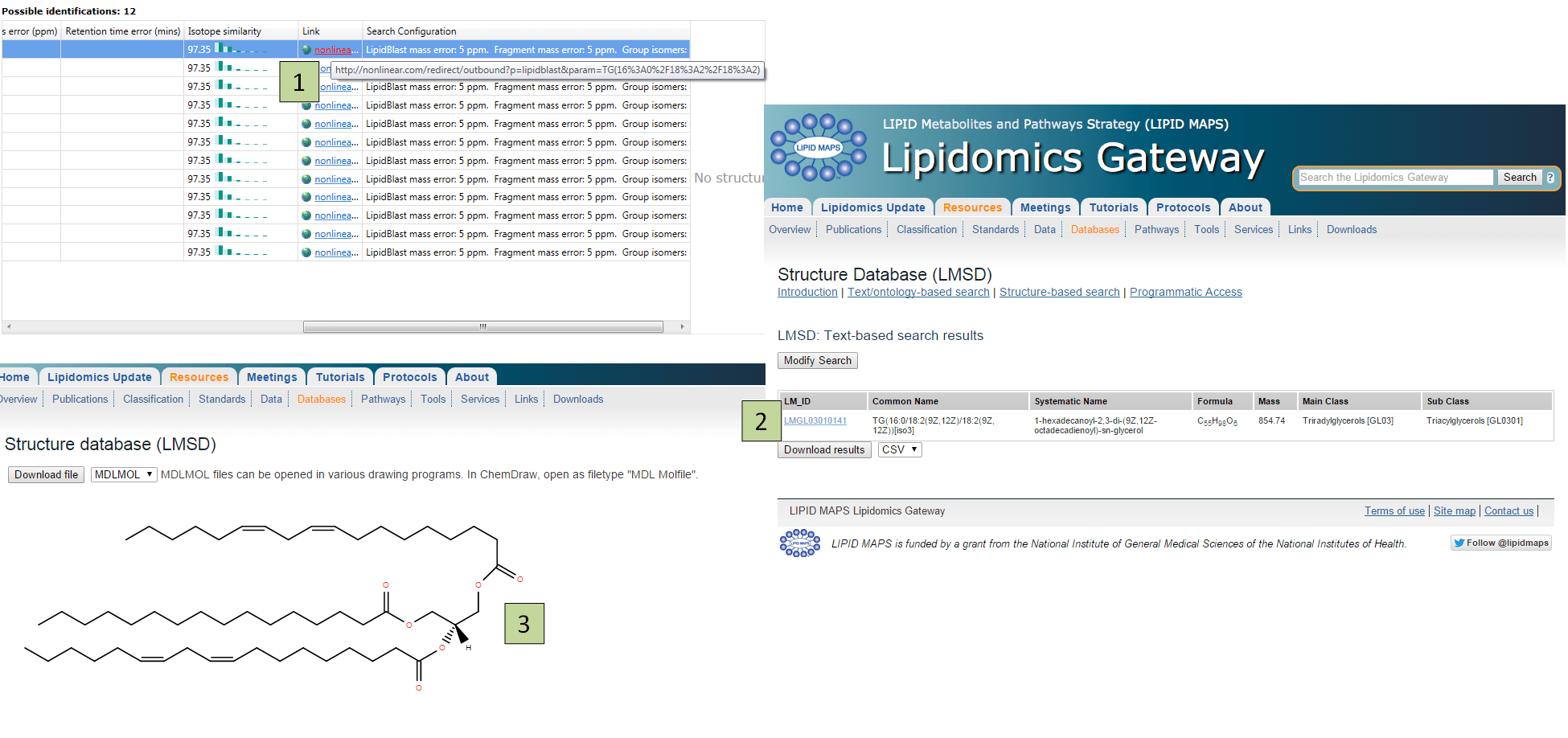
Following the link for an identification will take you to the appropriate LIPID MAPS entry, where you can view its structure.
References
- Kind et al., 2013: "LipidBlast in silico tandem mass spectrometry database for lipid identification". Nature Methods 10 (8), 755-8. DOI: 10.1038/nmeth.2551.